
Download


Protocol
Use of retroviral-mediated gene transfer to deliver and test function of chimeric antigen receptors in human T-cells
Ana C. Parente-Pereira1, Scott Wilkie2, Sjoukje J.C. van der Stegen1, David M. Davies3, John Maher1,4,5,*
1King’s College London, Department of Research Oncology, Guy’s Hospital Campus, Great Maze Pond, London SE1 9RT, UK
2Drug Discovery Unit, James Black Centre, University of Dundee, College of Life Sciences, Dow Street, Dundee, DD1 5EH, UK
3University College London Cancer Institute, London, WC1E 6DD, UK
4Department of Immunology, Barnet and Chase Farm NHS Trust, Barnet, Hertfordshire, EN5 3DJ, UK
5Department of Allergy and Clinical Immunology, King’s College Hospital NHS Foundation Trust, Denmark Hill, London SE5 9RS, UK
2Drug Discovery Unit, James Black Centre, University of Dundee, College of Life Sciences, Dow Street, Dundee, DD1 5EH, UK
3University College London Cancer Institute, London, WC1E 6DD, UK
4Department of Immunology, Barnet and Chase Farm NHS Trust, Barnet, Hertfordshire, EN5 3DJ, UK
5Department of Allergy and Clinical Immunology, King’s College Hospital NHS Foundation Trust, Denmark Hill, London SE5 9RS, UK
*Correspondence to: John Maher, Telephone 0044 207 188 1468; FAX 0044 207 188 0919. E-mail address: john.maher@kcl.ac.uk
Competing Interests: The authors have declared that no competing interests exist.
Abbreviations used: CAR, chimeric antigen receptor; GALV, gibbon ape leukemia virus; PC5, Phycoerythrincyanin 5.1; VSV, vesicular stomatitis virus
Received July 27, 2014; Revision received August 28, 2014; Accepted August 30, 2014; Published September 14, 2014
Abstract Chimeric antigen receptors (CARs) are genetically delivered fusion molecules that elicit T-cell activation upon binding of a native cell surface molecule. These molecules can be used to generate a large number of memory and effector T-cells that are capable of recognizing and attacking tumor cells. Most commonly, stable CAR expression is achieved in T-cells using retroviral vectors. In the method described here, retroviral vectors are packaged in a two-step procedure. First, H29D human retroviral packaging cells (a derivative of 293 cells) are transfected with the vector of interest, which is packaged transiently in vesicular stomatitis virus (VSV) G pseudotyped particles. These particles are used to deliver the vector to PG13 cells, which achieve stable packaging of gibbon ape leukemia virus (GALV)-pseudotyped particles that are suitable for infection of human T-cells. The key advantage of the method reported here is that it robustly generates polyclonal PG13 cells that are 100% positive for the vector of interest. This means that efficient gene transfer may be repeatedly achieved without the need to clone individual PG13 cells for experimental pre-clinical testing. To achieve T-cell transduction, cells must first be activated using a non-specific mitogen. Phytohemagglutinin (PHA) provides an economic and robust stimulus to achieve this. After 48-72 h, activated T-cells and virus-conditioned medium are mixed in RetroNectin-coated plasticware, which enhances transduction efficiency. Transduced cells are analyzed for gene transfer efficiency by flow cytometry 48 h following transduction and may then be tested in several assays to evaluate CAR function, including target-dependent cytotoxicity, cytokine production and proliferation.
Keywords: retroviral; transduction; T-cell; chimeric antigen receptor
Introduction
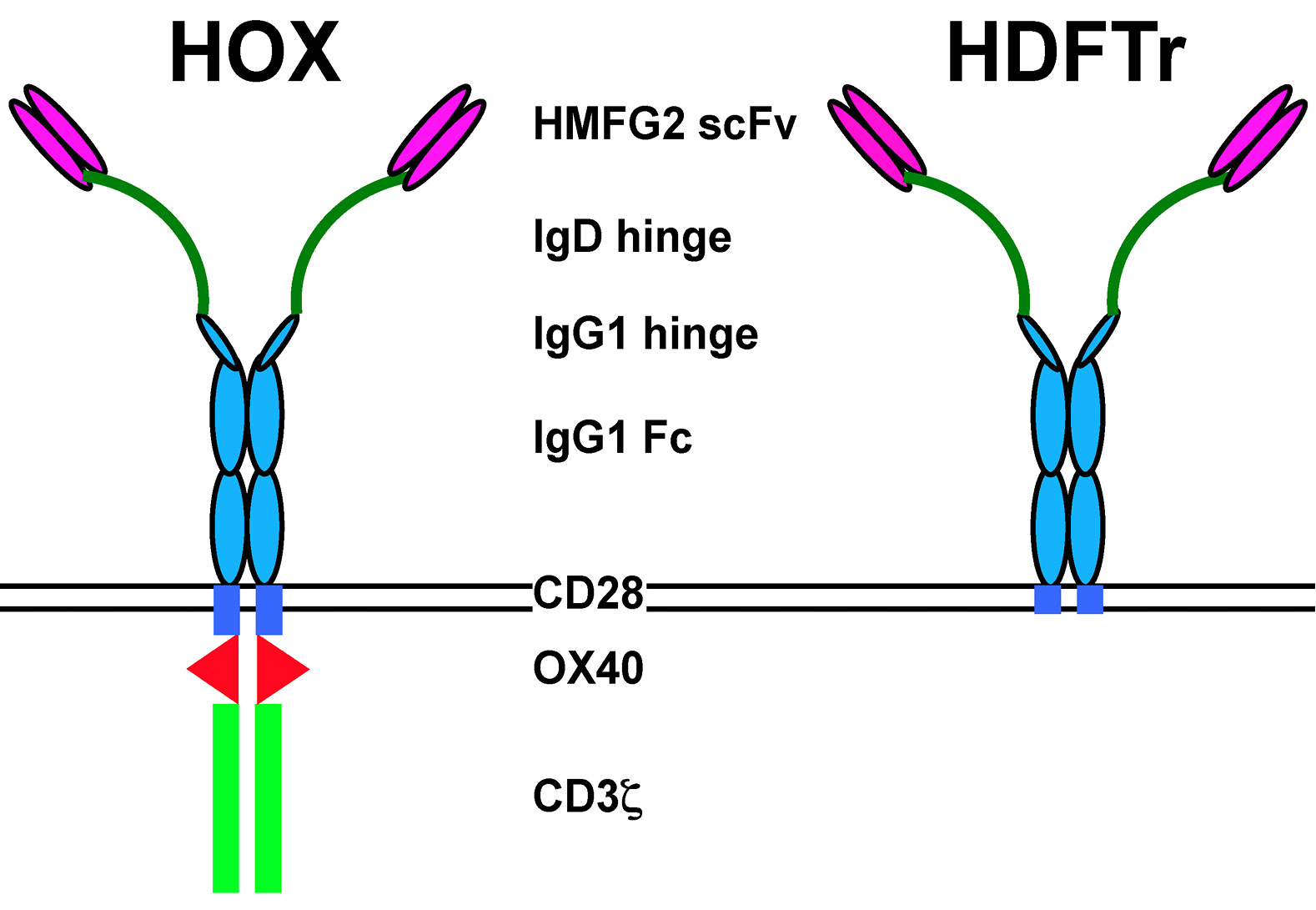
Chimeric antigen receptors (CARs) are bespoke fusion molecules that couple the binding of a selected cell surface target molecule to the delivery of a tailored T-cell activating signal. Developed over 25 years ago, CAR technology has advanced to the point that several clinical trials are now ongoing in which this technology is being tested in patients with diverse malignancies [1,2]. Recently, durable complete clinical remissions have been achieved in patients with B-cell malignancy and neuroblastoma using this technology [3-7]. Signaling by CARs is most commonly provided by a fused endodomain in which the CD3z subunit of the TCR/CD3 complex (signal 1) is combined with one or more co-stimulatory elements, including CD28, OX40 or 4-1BB (signal 2). Most commonly, the CAR ectodomain consists of a single chain antibody fragment. Figure 1 shows a typical example of such a composite CAR, named Human milk fat globulin 2 (HMFG2)-derived CAR containing OX40 (HOX) and targeted against the MUC1 mucin [8]. Alternatively, CARs may be targeted using a ligand (e.g. interleukin (IL)-13 to target IL-13 receptor-a2 or CD27 to target CD70) [9,10] or chimeric ligand with multiple target specificities (e.g. the T1E peptide – to target several ErbB dimers) [11].
Most commonly, CAR expression is achieved in human T-cells using gamma retroviral vectors. These vectors achieve stable transgene expression but have not been linked to clinically significant genotoxicity when expressed over many years in T-cells [12]. Retroviral vectors also provide a convenient system for pre-clinical testing and refinement of CAR-based immunotherapy. Stable retroviral packaging master cell banks based upon the PG13 cell line are widely used to achieve gene transfer in the clinical evaluation of this technology. Here, we describe a practical method to transduce activated human T-cells with CAR-encoding retroviral vectors and illustrate how transgene expression and function may subsequently be evaluated in vitro.
A two-step method is described for production of virus-like particles, involving the sequential use of H29D and PG13 cells. The H29D retroviral packaging cell line is derived from adenovirus 5-transformed 293 cells that have been engineered to express retroviral gag-pol polyprotein constitutively [13]. These cells also produce the vesicular stomatitis virus (VSV) G protein in a manner that is suppressed by tetracycline. Since VSV G is toxic, H29D cells are routinely propagated in the presence of tetracycline and this is removed when viral packaging is required. Viral particles derived from H29D cells are used to produce a stable PG13 retroviral packaging cell line. PG13 cells produce retroviral particles with a gibbon ape leukemia virus pseudotype (GALV), suitable for stable transduction of activated human T-cells [14]. Virus produced by H29D cells is less amenable for gene transfer into human T-cells since gene transfer is less efficient and compromised by pseudo-transduction [15].
The method described here provides an alternative to gene transfer using the Sleeping Beauty transposon system [16]. While clinical experience using transposon-based methods is limited, retroviral vectors have proven track record of safety when used in the clinical engineering of T-cells [12]. Furthermore, transposon-based systems generally require the electroporation of T-cells and are frequently less efficient than retroviral systems. Use of the PG13 cell line allows the generation of a stable retroviral packaging cell line for repeated pre-clinical testing in human T-cells and is amenable to the generation of master and working cell banks for clinical use. The key advantage of the method reported here is that it robustly generates polyclonal PG13 cells that are 100% positive for the vector of interest. This means that efficient gene transfer may be achieved repeatedly without the need to clone individual PG13 cells for pre-clinical testing. Thus, our protocol expands upon previously reported methods to achieve retroviral transduction of human T-cells [15,17]. Furthermore, if ecotropic packaging cells are substituted for PG13 cells, the method may be adapted to ensure efficient transduction of murine T-cells [18], including naïve and central memory sub-types [19].
Materials
- Aldesleukin (Human recombinant interleukin-2) (Novartis)
- Antibiotic antimycotic solution (Sigma-Aldrich, A5955)
- BT20 (Cancer Research UK)
- Butterfly needle and Luer adaptor 21G x 0.75 inches (Greiner Bio-one, cat # 450066)
- Calcium phosphate transfection kit (Sigma-Aldrich, cat # CAPHOS-1KT)
- C412 Centrifuge (Jouan)
- CD4-FITC conjugated (Miltenyi Biotec, cat # 130-080-501)
- CD8-PC5 conjugated (Beckman Coulter, cat # A07758)
- Citrate-dextrose solution (Sigma-Aldrich, cat # C3821)
- Crystal violet (Sigma-Aldrich, cat # C3886)
- DMEM with glucose (Lonza, cat # Lonz12-614F/12)
- Fetal bovine serum (Autogen Bioclear)
- Fibronectin fragment (RetroNectin) (Takara BioEurope Clontech, cat # T100B)
- Ficoll-paque PLUS (GE Healthcare, cat # 17-1440-02)
- G418 (Sigma-Aldrich, cat # A1720)
- Glutamax (Invitrogen (Life Technologies, cat # 35050038)
- Human AB serum (male) (Sigma-Aldrich, cat # H4522-100ml)
- Human IL-2 ELISA Ready-Set-Go (eBioscience, cat # 88-7026-88)
- Human interferon (IFN)-g ELISA Ready-Set-Go (eBioscience, cat # 88-7316-88)
- MDA-MB-231 (Cancer Research UK)
- MDA-MB-435 (Cancer Research UK)
- Methanol (Fisher Scientific, cat # M/4000/17)
- MUC1 24mer peptide: N-terminal biotinylated TAPPAHGVTSAPDTRPAPGSTAPP (NeoMPS)
- PG13 cells (European Collection of Cell Cultures, cat # 95110215)
- Phosphate buffered saline (Gibco, cat # 10010-015)
- Phytohemagglutinin (Phaseolus vulgaris Leukoagglutinin PHA-L) (Sigma-Aldrich, cat # L 2769)
- Polybrene (polymethobromide, hexadimethrine bromide or hexadimethrine bromide) (Sigma-Aldrich, cat # H9268)
- Puromycin (Sigma-Aldrich, cat # P9620)
- RPMI 1640 without l-glutamine (Lonza, cat # 733-1690)
- Six well tissue culture-treated plates (Greiner Labortechnik, cat # 700-1425)
- Six well non tissue culture-treated plates (Becton Dickinson, cat # 734-0948)
- Streptavidin – phycoerythrin conjugated (Invitrogen (Life Technologies), cat # S866)
- Syringe polypropylene 50 ml with Luer Lok (Fisher Scientific, cat # SZR-150-080U)
- T47D (Cancer Research UK)
- Tetracycline (Cell culture grade; water soluble at 10 mg/ml. Store as frozen aliquots) (Sigma-Aldrich, cat # T7660)
- Trypan blue solution, 0.4% in phosphate buffered saline (Fisher Scientific Ltd, cat # HYC-044-010D)
- Thiazolyl blue tetrazolium bromide, 98% (Sigma-Aldrich, cat # M2128)
- Tubes – conical centrifuge tubes 50 ml (PAA Laboratories, cat # PAA50050)
- Tubes – polystyrene 12x75 mm 5 ml with cap (Becton Dickinson, cat # 352054)
- Twenty four well tissue culture treated plates (PAA Laboratories, cat # PAA32024X)
Procedure
A schematic representation of the protocol is shown in the Graphical Abstract (available online). All steps are carried out under sterile conditions, working in a class II laminar flow cabinet.
1.Transient virus production using H29D cells.
1.1.Maintain H29D cells (a gift of Dr. Michel Sadelain, Memorial Sloan Kettering Cancer Center, NY) in 6 well plates containing 4 ml of DMEM + 10% FBS + antibiotic/antimycotic solution + glutamax (D10 medium). Medium is supplemented with 2 µg/mL tetracycline (to maintain repression of VSV G expression), 0.3 mg/ml G418 (to maintain gag-pol expression) and 2 µg/ml puromycin (to maintain tetracycline-regulated VSV G expression). Cells are passaged when they reach 90% confluence by trypsinization.
1.2.Achieve CAR expression using the SFG gamma retroviral vector [20] (a gift of Dr. Michel Sadelain). Transgene expression is driven by the retroviral long terminal repeat.
1.3.On the day of transfection (day 1), select a well containing H29D cells that is 80-90% confluent. Transfection of H29D is optimally achieved if cells are about 80% confluent. Remove medium and replace with 4 ml DMEM + 10% FBS (e.g. without additional tetracycline, G418 or puromycin). Return to the incubator (37°C and 5% CO2) for at least 3 h.
1.4.Transfect H29D cells with the plasmid containing the SFG vector encoding the CAR of interest (e.g. HOX, Fig. 1) using Calcium Phosphate Transfection Kit, as described by the manufacturer.
1.5.After 24 h, replace the medium with fresh D10 (e.g. lacking any supplemental G418, puromycin or tetracycline). Inspect the cells and medium daily and replace the medium before day 5 if the medium turns yellow.
1.6.Harvest supernatants daily from day 5 until day 8, at which point VSV G-mediated syncytialization of H29D cells should be visible using the inverted microscope. Harvested supernatants may be used to infect target cells (see below) or alternatively may be snap frozen in an ethanol bath.
1.7.Replace harvested supernatants daily with 4 ml fresh D10 media. By day 8, H29D cells generally appear highly syncytialized and cultures deteriorate. H29D cells cannot be propagated for longer periods in the absence of tetracycline.
Figure 1. The MUC1-specific CAR, HOX. In HOX, reactivity with tumor-associated glycoforms of MUC1 is conferred by a single chain antibody fragment (scFv) derived from the HMFG2 hybridoma (8). To overcome MUC1-imposed steric hindrance, a flexible elongated hinge has been introduced derived from human IgD. CAR expression has been stabilized using IgG1 Fc. Signaling is provided by a composite endodomain in which CD28 and OX40 modules are placed upstream of CD3ζ. The HDFTr CAR is a matched control in which the endodomain has been truncated to render it inactive (8). HOX: Human milk fat globulin 2 (HMFG2)-derived CAR containing OX40. HDFTr: HMFG2-IgD-IgGFc-truncated CAR.
2.Preparation of PG13 retroviral packaging cells.
2.1.Propagate PG13 cells in 6 well plates containing 4 ml of D10 medium at 37°C and 5% CO2.
2.2.Ensure that PG13 cells are approximately 30% confluent and evenly dispersed throughout the well in order to achieve productive infection by H29D-derived viral particles.
2.3.Replace medium with 4 ml of H29D-derived viral particles, harvested as described in 1.6.
2.4.After addition of virus-containing medium, add polybrene (8 µg/mL). Mix thoroughly but gently and incubate cells at 37°C and 5% CO2.
2.5.After 24 h, replace medium with 4 ml D10 and incubate cells at 37°C and 5% CO2.
2.6.Assess efficiency of gene transfer from H29D supernatant to PG13 cells 24-48 h after transduction by flow cytometry.
2.6.1.Briefly, stain cells with a specific antibody (or other detection reagent) directed against the CAR. This will recognize an epitope contained within the CAR extracellular domain and is dependent upon the CAR under study. Incubate on ice for 20 min followed by two washes using PBS.
2.6.2.If the CAR-specific antibody is not directly conjugated with an appropriate fluorochrome, perform an additional staining step with a conjugated secondary antibody. Incubate samples on ice for 20 min and wash twice with PBS.
2.6.3.Analyze stained cells by flow cytometry. It is necessary to have appropriate compensation controls, as dictated by the flow cytometer in use.
2.6.4.Ensure that the PG13 cell line generated as described above is close to 100% positive for cell surface expression of the CAR of interest. Typically, best results are obtained with supernatants harvested on days 5-7 after transfection of H29D cells.
3.Transduction of activated human T-cells
3.1.Collect blood (45 ml) using a 21-gauge butterfly needle into a 50 ml syringe. Blood should be collected by certified personnel following Health Insurance Portability and Accountability Act (HIPAA) protocols.
3.2.Transfer harvested blood immediately after collection to anticoagulant (5 ml citrate dextrose solution) in a 50 ml Falcon tube.
3.3.Transfer Ficoll-Paque (15 ml) into each of two 50 ml conical centrifuge tubes.
3.4.Tilt the Ficoll-containing centrifuge tube as close to horizontal as possible. Draw up anticoagulated blood into a pipette and gently add to the side of the tube, aiming not to disrupt the interface between the blood and ficoll. As blood is added, the tube is gradually brought to the vertical position.
3.5.Repeat this process for both ficoll tubes. Some settling of red cells through the ficoll layer may be observed, particularly in the first tube and is not of concern.
3.6.Centrifuge the tubes at 500 g for 25 min. Ensure the acceleration and deceleration settings of the centrifuge are at zero. The centrifuge should not be refrigerated.
3.7.Transfer the peripheral blood mononuclear cell (PBMC) layer, present at the interface between the Ficoll-Paque and the plasma, into a fresh 50 ml tube using a Pasteur pipette. Dilute the cells to a final volume of 50 ml in PBS.
3.7.1.Centrifuge at room temperature for 10 min at 370 g.
3.8.Aspirate the supernatant and re-suspend the cell pellet in 50 ml PBS.
3.8.1.Centrifuge at room temperature for 10 min at 270 g.
3.9.Aspirate the supernatant and re-suspend the cell pellet in RPMI 1640 + 10% human AB serum + antibiotic/antimycotic solution + glutamax (R10 medium).
3.10.Plate the cells at a density of 3 × 106 cells/ml in a six well tissue culture treated plate (4 ml per well).
3.11.Add phytohemagglutinin (PHA; 5 µg/ml) to activate T-cells and incubate at 37°C and 5% CO2.
Note: Alternatively to PHA, T-cells may be activated with CD3 + CD28-coated paramagnetic beads (e.g. Cell Expander Dynabeads (cat # 11141D), Invitrogen (Life Technologies), as indicated by the manufacturers).
3.12.Add IL-2 (100 U/ml) 24 h prior to performing gene transfer. Gene transfer may be conducted either 48 or 72 h after T-cell activation with PHA. Lower concentrations of IL-2 may be used for this purpose.
3.13.One day prior to performing gene transfer, prepare RetroNectin plates as described by the manufacturer. Split confluent PG13 retroviral packaging cells 1 in 2 by trypsinization. Each of the resulting cell aliquots is plated in 4 ml fresh D10. After incubation at 37°C and 5% CO2 for 24 h, PG13 cells generally reach confluence and medium can be harvested as a source of viral vector for T-cell gene transfer. Prepare one well containing 4 ml viral vector conditioned medium for each transduction of 1 × 106 PBMC.
3.14.On the day of gene transfer, remove unbound RetroNectin from the non-tissue culture treated six well plate.
3.15.Do not allow RetroNectin-coated wells to dry out. Following removal of unbound RetroNectin, transfer 4 ml of PG13 viral conditioned medium promptly to each well of the RetroNectin-coated plate and leave in the flow cabinet at room temperature during the minutes required to collect activated PBMC. This order is recommended since PBMC should be kept in the incubator whenever possible.
3.16.Collect activated PBMC in a 50 ml conical tube and count viable cells by trypan exclusion using a hemocytometer. Do not centrifuge the cells as conditioned medium is likely to facilitate gene transfer efficiency.
3.17.Determine the volume that contains 1 million activated PBMC (mainly T-cells) and, after careful mixing by inversion, transfer this volume to each well of the RetroNectin-coated plate containing viral conditioned medium.
3.18.Add IL-2 (100 U/ml) to each well. This is most conveniently achieved by adding the appropriate amount of IL-2 to the activated PBMC stock and then distributing the cytokine-supplemented cells to the RetroNectin-coated plate.
3.19.Centrifuge plates at 50 g for 1 h at room temperature. This step is believed to facilitate gene transfer. However, transduction may still be achieved if this step is omitted. Incubate cultures at 37°C and 5% CO2.
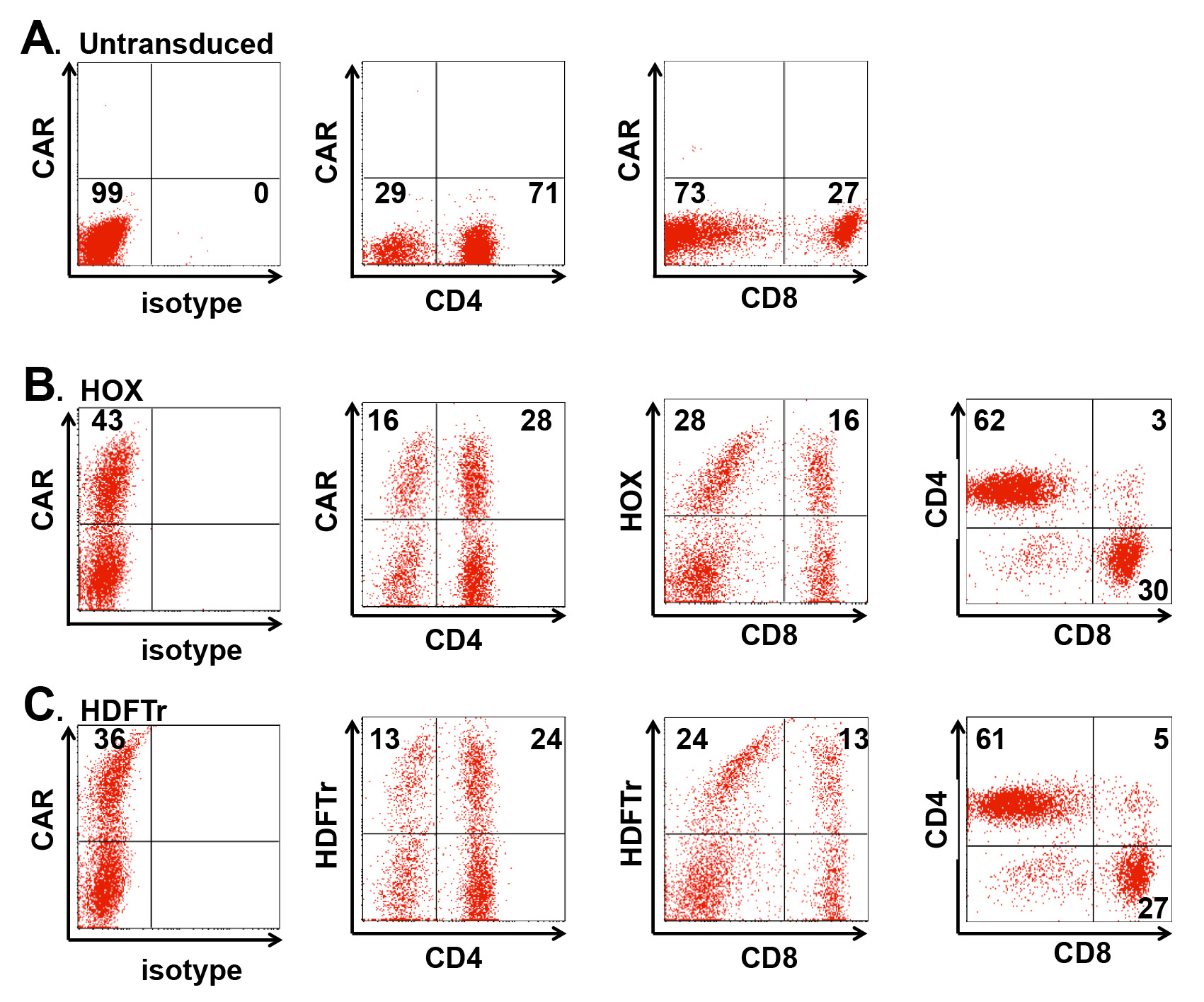
Figure 2. Flow cytometric assessment of gene transfer efficiency. T-cells were transduced with SFG HOX (MUC1-specific CAR) and SFG HDFTr (matched endodomain truncated control CAR). A. Isotype and compensation controls. To detect CAR expression on the cell surface, cells are incubated on ice with a biotinylated 24mer peptide (produced in house) containing one copy of the MUC1 epitope recognized by the HOX (B) or HDFTr (C) control CARs. After washing, cells are then incubated with phycoerythrin-coated streptavidin, together with a fluorescein-conjugated CD4 antibody and a Phycoerythrincyanin 5.1 (PC5)-conjugated CD8 antibody prior to analysis by flow cytometry. Quadrant markers are set using untransduced T-cells. Percentage of events indicate the proportion of transduced CD4+ and CD8+ T-cells present, expressed as a percentage of all T-cells present in the culture.
4.Propagation of retrovirus-transduced T-cells and examination of CAR expression
4.1.Two days after gene transfer, analyze transgene expression by flow cytometry as indicated below. Typically, a transduction efficiency in excess of 35% of T-cells is attained (Fig. 2), incorporating both central memory and effector memory T-cell types [21].
4.1.1.Incubate transduced and untransduced control T-cells (about 2 x 105 cells per tube) with CAR detection reagent on ice. In the example shown (Fig. 2), 1 µg of MUC1 24mer peptide (CAR detection reagent) was added for 30 min, to enable the detection of MUC1-specific HOX and control HDFTr CARs.
4.1.2.After washing with cold PBS (4°C), 1 µl of Streptavidin-PE conjugate was added and samples were placed on ice. Proceed immediately to step 4.1.3.
4.1.3.To determine relative CAR expression in T-cell subsets, incubate cells with antibodies that bind CD4 and/or CD8. 10 µl of CD4-FITC and 10 µl of CD8-PC5 were added for 15 min on ice. Matched control tubes should be set up in which conjugated isotype control antibodies are incubated similarly. After incubation, all tubes were washed with cold PBS (4°C).
4.1.4.Compensation settings need to be adjusted prior to analysis of “triple stained” cells by flow cytometry. Set up three separate tubes of HOX- (or HDFTr-) transduced T-cells and incubate as described above with either: (i) MUC1 24mer peptide followed by Streptavidin-PE; (ii) CD4-FITC or (iii) CD8-PC5.
4.1.5.After washing of tubes with cold PBS (4°C), these samples are used to adjust compensation settings, as illustrated in the example shown in Figure 2.
4.1.6.Analyze triple stained tubes by multicolor flow cytometry. Use isotype control settings (for CD4 and CD8) or untransduced control settings (for CAR detection reagents) to set quadrant markers on fluorescence emission dot plots. This enables the determination of the percentage transduction efficiency of individual T-cells subsets (Fig. 2).
4.1.7.If an unconjugated primary antibody followed by conjugated secondary antibody is used to detect CAR expression, accuracy of detection of other markers may be compromised by incomplete blockade of the secondary reagent. To avoid this: (i) Perform steps 4.1.1 to 4.1.5 as above, adding primary and secondary antibody reagents sequentially and washing as indicated. (ii) Next, add mouse serum (10 µl of ١/٥٠ dilution) to block unoccupied antigen-combining sites in the secondary fluorochrome-conjugated antibody. Incubate for 20 min on ice. There is no need to wash after this step. (iii) Add fluorochrome conjugated CD4 and/or CD8 antibodies. Incubate for 20 min on ice. Wash using cold PBS (4°C).
4.2.Propagate transduced T-cells at 37°C and 5% CO2 in R10 medium with IL-2 (100 U/ml) until required for experimental purposes. Add fresh medium and cytokine three times per week or more frequently if a change in medium color occurs, consistent with acidification.
5.Testing of CAR functionality in retrovirus-transduced T-cell cultures. Test functionality by performing co-culture experiments in which CAR-engineered T-cells are placed on paired adherent target monolayers that are discordant for expression of the antigen of interest. An example is NIH/3T3 fibroblasts that lack the target antigen or which have been genetically modified to express this molecule [22]. Additional control cultures should include T-cells that are untransduced and/or engineered to express CARs that lack ectodomain or endodomain elements e.g. the truncated control CAR, HMFG2-IgD-IgGFc-truncated CAR (HDFTr, Fig. 1). Quantify T-cell activation by measurement of target cell destruction, cytokine production and/or T-cell proliferation.
5.1.Culture target cell monolayers to confluence in 24 well tissue culture plates containing the appropriate medium (generally D10 is sufficient).
5.2.Centrifuge engineered T-cells for 5 min at 200 g and re-suspend at 1 × 106 cells/ml in R10 without exogenous cytokine. Where two CARs are being compared, it may be appropriate to add untransduced T-cells to some cultures in order to equalize the proportion of transduced cells present.
5.3.Remove medium from target monolayer cultures. Add 1 ml of T-cells (1 × 106 cells).
5.4.After 24 h, remove supernatant for measurement of cytokine content (e.g. IL-2, IFN-g). Cytokine production is a useful and objective method to quantify T-cell activation upon encounter with tumor cells. Supernatants are analyzed according to manufacturers instructions which may require dilution in order that optical density values lie on the linear portion of the standard curve.
5.5.Quantify target cell destruction using an MTT (3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazolium bromide; thiazolyl blue) assay, as described by the manufacturer.
5.6.Alternatively, visualize monolayer destruction by crystal violet staining. It is essential to prepare a well containing an adherent tumor cell monolayer alone as a control (Fig. 3).
5.6.1.Aspirate medium carefully after completion of the T-cell monolayer co-cultivation.
5.6.2.Wash wells gently with 500 µl PBS and then fix using 500 µl of ice-cold methanol, incubated at – 20°C for a minimum of 10 min.
5.6.3.Aspirate methanol and submerge each well in crystal violet (0.5% solution in 25% methanol, stored at room temperature). Incubate the plate at room temperature for 5 min.
5.6.4.Aspirate crystal violet and wash plates by gentle submersion in water to remove excess dye.
5.6.5.Dry plates at room temperature overnight.
5.6.6.Capture light microscopy images using an inverted microscope with appropriate software.
5.7.Quantify T-cell proliferation as follows (Fig. 4)
5.8.Establish T-cell/target cell co-cultivations as described in 5.1 – 5.3.
5.8.1.Add IL-2 (100 U/ml) after 24 h and every 2-3 days thereafter, together with R10 medium, as dictated by the appearance of the cultures.
5.8.2.Transfer cultures to a 6 well plate when volume exceeds 1.5 ml.
5.8.3.Determine cell number by trypan exclusion at the appropriate interval, generally every 7 days.
5.8.4.Re-evaluate the proportion of CAR+ T-cells present in the culture by flow cytometry, generally every 7 days.
5.8.5.Re-stimulate cultures if all target cells have been destroyed by the CAR-engineered T-cells. Generally, this is performed after 7 days and is achieved by placing 1 × 106 T-cells on a fresh target cell monolayer in a 24 well plate.
5.8.6.Conduct periodic re-stimulation in this manner until cultures are no longer capable of expanding.
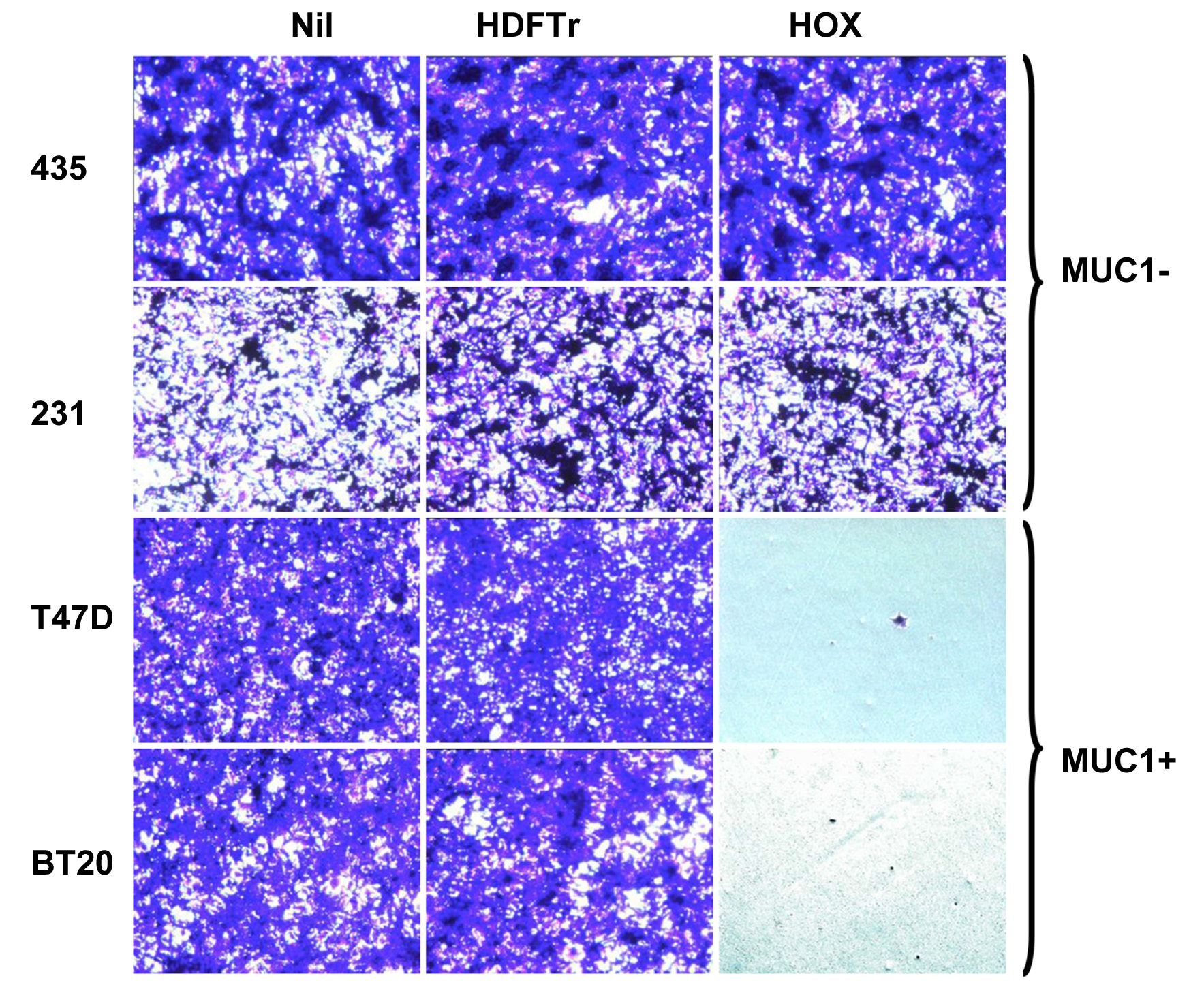
Figure 3. Crystal violet monolayer destruction assay. Engineered T-cells (1 × 106 cells) were co-cultivated for 24 h with confluent monolayers of the indicated MUC1- (MDA-MB-435; MDA-MB-231) or MUC1+ (T47D; BT20) breast tumor cell lines, cultured in 24-well plates. Non-adherent cells were then removed and, after fixation, residual tumor monolayers were stained using crystal violet. “Nil” indicates monolayers that were not co-cultivated with T-cells.
Figure 4. Appearance of T-cell monolayer co-cultures after 72 h. Co-cultures were established as described in Figure 3 in which T-cells were incubated with MUC1+ T47D cells. “Nil” indicates monolayer alone. Note that the monolayer remains intact when HDFTr+ T-cells are added. By contrast, HOX+ T-cells have destroyed the T47D monolayer and form large clusters of proliferating activated T-cells, admixed with dead tumor cells.
Anticipated Results
The protocol described here is a robust methodology to achieve T-cell transduction. The most critical parameters for successful gene transfer are robustly activated T-cells and use of freshly generated retroviral supernatants from validated PG13 cell stocks (Table 1). HOX is a CAR that can bind several tumor-associated glycoforms of the MUC1 mucin, commonly found in breast carcinomas, but which are under-represented in normal tissues (Fig. 1) [8,23]. HDFTr is a matched control CAR in which the endodomain has been truncated, rendering it functionally inactive (Fig. 1) [8]. Figure 2 shows a representative example of transduction efficiency achieved when these CARs were expressed using the SFG gamma retroviral vector, delivered to human T-cells using the protocol described above. Note that both CD4+ and CD8+ T-cells are transduced at comparable efficiencies, allowing for the relative proportion of CD4+ and CD8+ T-cells that were present in these PHA-activated T-cell cultures. Furthermore, use of the SFG vector generally achieves high-level transgene expression in human T-cells (Fig. 2). Functional studies can generally provide a robust discrimination between CAR-transduced and control T-cell populations. However, with some weakly active CARs, this may be less evident [8]. Assays based upon MTT reduction and crystal violet staining are useful methods to quantify and visualize T-cell killing respectively. However, both assays are limited to target cells that are adherent. Figure 3 shows an experiment in which monolayer destruction was visualized using crystal violet staining. Note that HOX+ but not control HDFTr+ T-cells cause the selective elimination of breast cancer monolayers that naturally express the MUC1 mucin. MUC1-dependent selective tumor cell killing by HOX+ CAR T-cells is also seen when MUC1 is ectopically expressed in tumor cells such as MDA-MB-435 [8]. When CAR+ T-cells undergo antigen-driven activation, they also form large clusters within the wells and undergo proliferation (Fig. 4). In our experience however, cytokine production remains the most useful discriminator of function between CARs under study. When CAR+ T-cells are activated, they generally produce high levels of IFN-γ, which correlates with destruction of the target cells [8,24]. Additionally, effective second- and third-generation CARs (which contain one or two co-stimulatory modules respectively) generally enable T-cells to produce greater quantities of IL-2 and to proliferate more robustly.
Table 1. Troubleshooting table.
Acknowledgements
This research was supported by the Breast Cancer Campaign, the Experimental Cancer Medicine Centre at King's College London and by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. Support was also provided by the US Department of Defense under contract number W81XWH-09-1-0096, British Lung Foundation and Association for International Cancer Research.
References
Maher J (2012) Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol 2012: 278093. doi: 10.5402/2012/278093. [View Article] [PubMed] [Google Scholar]
Maher J (2014) Clinical immunotherapy of B-cell malignancy using CD19-targeted CAR T-cells. Curr Gene Ther 14: 35-43. [PubMed] [Google Scholar]
Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, et al. (2008) Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14: 1264-1270. doi: 10.1038/nm.1882. [View Article] [PubMed] [Google Scholar]
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, et al. (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118: 6050-6056. doi: 10.1182/blood-2011-05-354449. [View Article] [PubMed] [Google Scholar]
Porter DL, Levine BL, Kalos M, Bagg A (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725-733. doi: 10.1056/NEJMoa1215134. [View Article] [PubMed] [Google Scholar]
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, et al. (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3: 95-73. doi: 10.1126/scitranslmed.3002842. [View Article] [PubMed] [Google Scholar]
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, et al. (2013) CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5: 177-138. doi: 10.1126/scitranslmed.3005930. [View Article] [PubMed] [Google Scholar]
Wilkie S, Picco G, Foster J, Davies DM, Julien S, et al. (2008) Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol 180: 4901-4909. doi: 10.4049/?jimmunol.180.7.4901. [View Article] [PubMed] [Google Scholar]
Kahlon KS, Brown C, Cooper LJN, Raubitschek A, Forman SJ, et al. (2004) Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res 64: 9160-9166. doi: 10.1158/0008-5472.CAN-04-0454. [View Article] [PubMed] [Google Scholar]
Shaffer DR, Savoldo B, Yi Z, Chow KKH, Kakarla S, et al. (2011) T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood 117: 4304-4314. doi: 10.1182/blood-2010-04-278218. [View Article] [PubMed] [Google Scholar]
Davies DM, Foster J, Van Der Stegen, J.C. (Sjoukje) , Parente-Pereira AC, Chiapero-Stanke L, et al. (2012) Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med 18: 565-576. doi: 10.2119/molmed.2011.00493. [View Article] [PubMed] [Google Scholar]
Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G (2012) Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4: 132-153. doi: 10.1126/scitranslmed.3003761. [View Article] [PubMed] [Google Scholar]
Ory DS, Neugeboren BA, Mulligan RC (1996) A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci U S A 93: 11400-11406. [PubMed] [Google Scholar]
Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C, et al. (1991) Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol 65: 2220-2224. [PubMed] [Google Scholar]
Gallardo HF, Tan C, Ory D, Sadelain M (1997) Recombinant retroviruses pseudotyped with the vesicular stomatitis virus G glycoprotein mediate both stable gene transfer and pseudotransduction in human peripheral blood lymphocytes. Blood 90: 952-957. [PubMed] [Google Scholar]
Huls MH, Figliola MJ, Dawson MJ, Olivares S, Kebriaei P, et al. (2013) Clinical application of Sleeping Beauty and artificial antigen presenting cells to genetically modify T cells from peripheral and umbilical cord blood. J Vis Exp 72: doi: 10.3791/50070. [View Article] [PubMed] [Google Scholar]
Rivière I, Gallardo HF, Hagani AB, Sadelain M (2000) Retroviral-mediated gene transfer in primary murine and human T-lymphocytes. Mol Biotechnol 15: 133-142. doi: 10.1385/MB:15:2:133. [View Article] [PubMed] [Google Scholar]
Zhong S, Malecek K, Perez-Garcia A, Krogsgaard M (2010) Retroviral transduction of T-cell receptors in mouse T-cells. J Vis Exp 44: doi: 10.3791/2307. [View Article] [PubMed] [Google Scholar]
Kerkar SP, Sanchez-Perez L, Yang S, Borman ZA, Muranski P, et al. (2011) Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J Immunother 34: 343-352. doi: 10.1097/CJI.0b013e3182187600. [View Article] [PubMed] [Google Scholar]
Rivière I, Brose K, Mulligan RC (1995) Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci U S A 92: 6733-6737. [PubMed] [Google Scholar]
Schuberth PC, Jakka G, Jensen SM, Wadle A, Gautschi F, et al. (2012) Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther 20: 386-395. doi: 10.1038/gt.2012.48. [View Article] [PubMed] [Google Scholar]
Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M (2002) Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 20: 70-75. doi: 10.1038/nbt0102-70. [View Article] [PubMed] [Google Scholar]
Maher J, Wilkie S (2009) CAR mechanics: driving T cells into the MUC of cancer. Cancer Res 69: 4559-4562. doi: 10.1158/0008-5472.CAN-09-0564. [View Article] [PubMed] [Google Scholar]
Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira ACP, Cleary S, et al. (2010) Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem 285: 25538-25544. doi: 10.1074/jbc.M110.127951. [View Article] [PubMed] [Google Scholar]